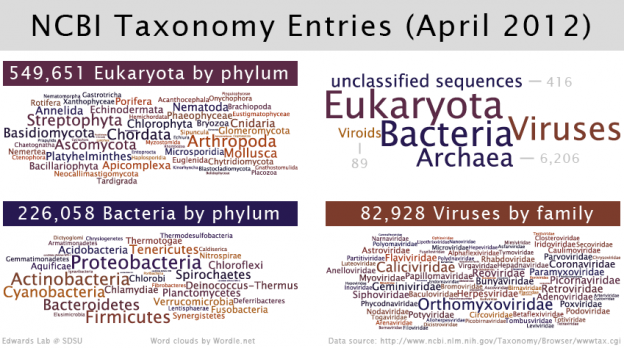
The NCBI Taxonomy database currently contains 865,348 different taxonomy entries that can be accessed using a unique identifier (the taxid). This unique identifier is an integer from 1 to 1,154,685 that can be used to access database entries at different taxonomic levels (kingdom, phylum, …). The graphic below summarizes the content of the NCBI Taxonomy database and highlights the phyla or families with the most entries. Most taxonomy entries by domain are for Eukaryota, followed by Bacteria and Viruses. Unclassified sequences include, for example, entries for metagenomes.

(Click on the image to see a larger version.)