We have a computational problem searching the Non-Redundant database, but we can solve that!
If you use MMSeqs2 to search the NR database, it needs about 1.75TB of RAM (that is approximate!). Our Flinders deepthought cluster has several nodes with 2.5TB RAM so at most you can run three concurrent jobs searching the database. However, database searches are completely independent: the scores you get for one pair of sequences are independent from all the other sequences in the database.
Note: That is true, but there is a caveat. The E-value of the search is dependent on the length of the query and the size of the database.
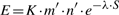
Therefore, the E-value that is reported is only approximate. There is a GitHub Issue to resolve this with MMSeqs2
Our solution is to split the database into smaller pieces, and then we can use those across more nodes of the cluster.
Download the NR database
Step 1, download the preformatted NR database using mmseqs2
mkdir --parents NR
mmseqs databases --threads 8 NR NR/NR (mktemp -d)
This will download the non-redundant database into the directory NR and the database will be called NR.
Split the database
Let’s split that database into many smaller chunks. From my tests it appears that 50 chunks requires about 50-60 GB RAM per compute, and 100 chunks requires about 25 GB RAM per compute.
CHUNKS=100
mkdir NR.split.$CHUNKS
cd NR.split.$CHUNKS
cut -f 1 ../NR/NR.index > ids.txt
split -n l/$CHUNKS --numeric-suffixes=1 --suffix-length=3 ids.txt
for N in $(seq 1 $CHUNKS); do
echo $N;
X="000$N";
X="x${X:(-3)}";
mkdir NR.$N
mmseqs createsubdb $X ../NR/NR NR.$N/NR;
mmseqs createsubdb $X ../NR/NR_h NR.$N/NR_h;
done
rm -f x???
(Hint: this works quite well as a slurm script!)
There are now 1 directory called NR.split.100 and within that there are 100 directories called NR.1, NR.2, NR.3, … NR.100.
Search against all the directories
We can now use a slurm array job to search against all of those:
#!/bin/bash
###############################################################
# #
# Search the NR using an array job. #
# #
# submit with this command: #
# #
# sbatch --array=1-100:1 ./nr_chunks.slurm #
# #
# Written by Rob Edwards. Good luck! #
# #
###############################################################
#SBATCH --job-name=NRmmseqs
#SBATCH --time=5-0
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=24
#SBATCH --mem=25G
#SBATCH -o mmseqsNRA_%a.out
#SBATCH -e mmseqsNRA_%a.err
## Memory requirement: for 100 chunks 25G (average from 72 searches: 16.3889, range: 12-20G)
set -euo pipefail
# change this to point to the output from `mmseqs createdb` on your data:
QUERY='mmseqs_formatted_data.db'
NR=$SLURM_ARRAY_TASK_ID
CHUNKS=100
mmseqs search --threads 24 $QUERY NR.split.$CHUNKS/NR.$NR/NR $QUERY.nr.$CHUNKS.$NR.db $(mktemp -d)
mmseqs convertalis --threads 24 $QUERY NR.split.$CHUNKS/NR.$NR/NR $QUERY.nr.$CHUNKS.$NR.db $QUERY.nr.$CHUNKS.$NR.m8
You can save this file as nr_chunks.slurm, edit the line with the QUERY location, and submit 100 MMSeqs2 jobs as an array job (using sbatch --array=1-100:1 ./nr_chunks.slurm). You now have 2,400 processors computing on your search, and each 24 processors is only consuming 25 GB RAM.
Note: I recommend that you adjust the number of threads, I am not sure if 24 is optimum!