PARTIE
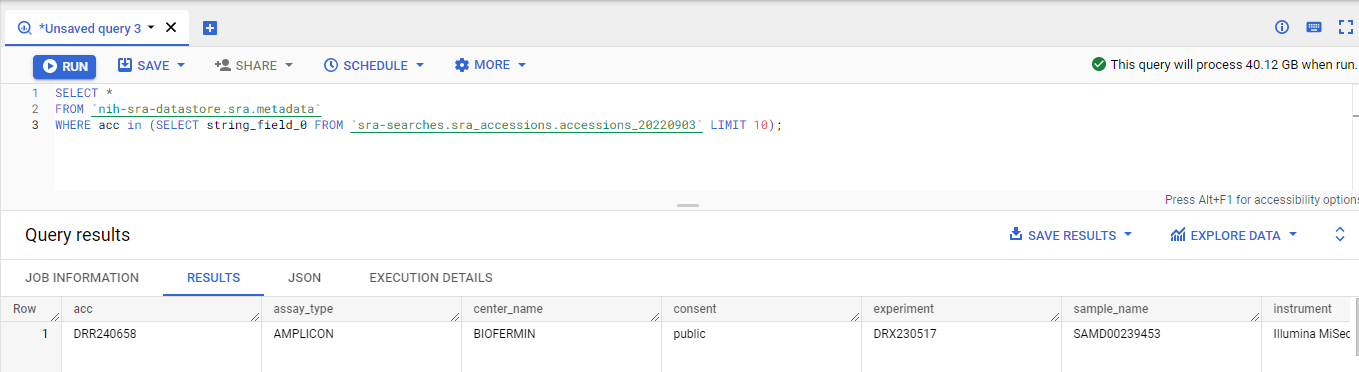
We have several projects that look for all the metagenomes in the cloud, and we have several ways of searching the SRA. Here, we’ll search for all the WGS metagenomes in the SRA using a Google Big Table query.
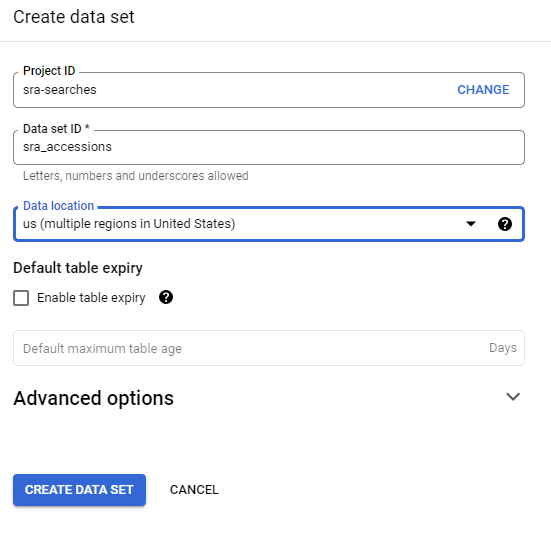
Log into Google Console
You’ll need to log into Google console and access a project or create a new one.
Use SQL to find the metagenome/microbiome/metatranscriptome results
We use temporary tables to store the two main searches: what are amplicon projects and what are metagenome/microbiome/metatranscriptome projects, and then we find the projects that are metagenomes:
create temp table AMPLICON(acc STRING) as select acc as amplicon from `nih-sra-datastore.sra.metadata` where assay_type = 'AMPLICON' or libraryselection = 'PCR';
create temp table METAGENOMES(acc STRING) as select acc from `nih-sra-datastore.sra.metadata` where librarysource = "METAGENOMIC" or librarysource = 'METATRANSCRIPTOMIC' or organism like "%microbiom%" OR organism like "%metagenom%" or organism like '%metatran%';
select acc from METAGENOMES where acc not in (select acc from AMPLICON);
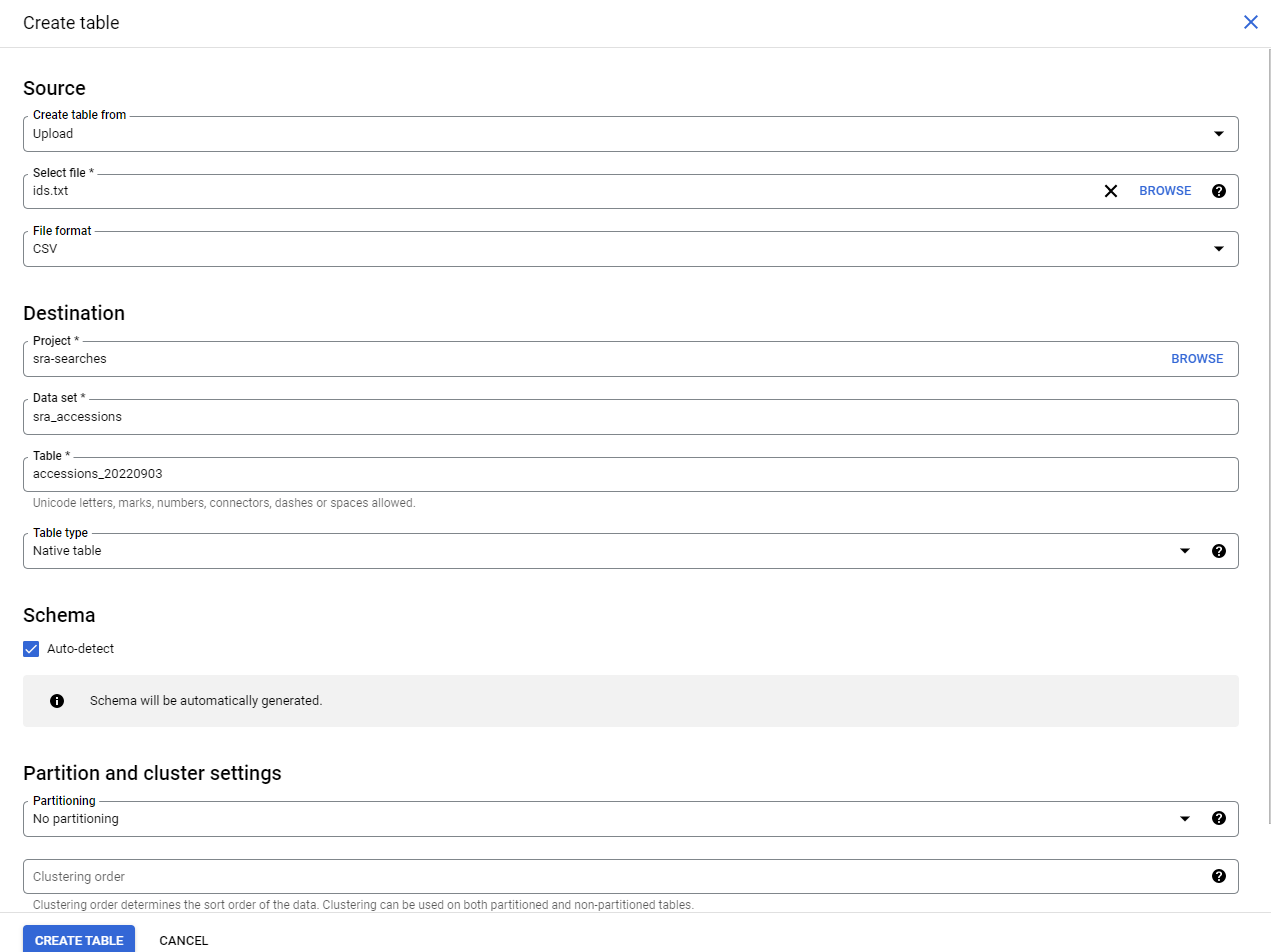
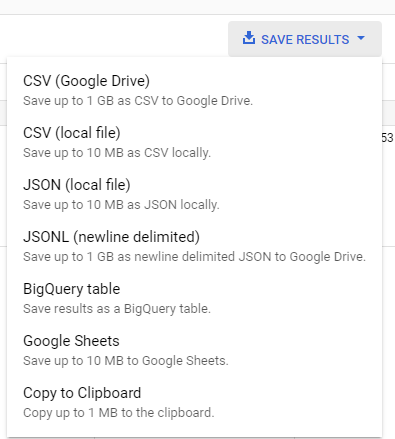
Then save that as a JSON file to Google Drive.
Use jq to parse the JSON file
This is probably overkill because we only have one attribute in our data.
jq -r '.acc' bq-results-20221006-054328-1665035790273.json > SRA-metagenomes.txt
Find all the information about all the sequences
We can edit the above SQL to get all the information about all the metagenomes. Basically, we just change the second select statement.
create temp table AMPLICON(acc STRING) as select acc as amplicon from `nih-sra-datastore.sra.metadata` where assay_type = 'AMPLICON' or libraryselection = 'PCR';
select * from `nih-sra-datastore.sra.metadata` where acc not in (select acc from AMPLICON) and (librarysource = "METAGENOMIC" or librarysource = 'METATRANSCRIPTOMIC' or organism like "%microbiom%" OR organism like "%metagenom%");
Note: In this query, the parenthesis are important to make sure we do the and and or in the right place.
Then you can export the data as a JSON Newline file to Google Drive.
Current results
At the moment, this returns 642,842 runs from the SRA
Some things we can’t find
- The old
study_typefield that we searched (usingstudy_type = "Metagenomics") does not appear to have mapped to bigtable. - THe old scientific name that we searched (using
sample.scientific_name like "%microbiom%" OR sample.scientific_name like "%metagenom%") does not appear to have mapped to bigtable.