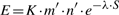We have a computational problem searching the Non-Redundant database, but we can solve that!
If you use MMSeqs2 to search the NR database, it needs about 1.75TB of RAM (that is approximate!). Our Flinders deepthought cluster has several nodes with 2.5TB RAM so at most you can run three concurrent jobs searching the database. However, database searches are completely independent: the scores you get for one pair of sequences are independent from all the other sequences in the database.
Note: That is true, but there is a caveat. The E-value of the search is dependent on the length of the query and the size of the database.
E value depends on the database size!
Therefore, the E-value that is reported is only approximate. There is a GitHub Issue to resolve this with MMSeqs2
Our solution is to split the database into smaller pieces, and then we can use those across more nodes of the cluster.
Download the NR database
Step 1, download the preformatted NR database using mmseqs2
mkdir --parents NR
mmseqs databases --threads 8 NR NR/NR (mktemp -d)
This will download the non-redundant database into the directory NR and the database will be called NR.
Split the database
Let’s split that database into many smaller chunks. From my tests it appears that 50 chunks requires about 50-60 GB RAM per compute, and 100 chunks requires about 25 GB RAM per compute.
CHUNKS=100
mkdir NR.split.$CHUNKS
cd NR.split.$CHUNKS
cut -f 1 ../NR/NR.index > ids.txt
split -n l/$CHUNKS --numeric-suffixes=1 --suffix-length=3 ids.txt
for N in $(seq 1 $CHUNKS); do
echo $N;
X="000$N";
X="x${X:(-3)}";
mkdir NR.$N
mmseqs createsubdb $X ../NR/NR NR.$N/NR;
mmseqs createsubdb $X ../NR/NR_h NR.$N/NR_h;
done
rm -f x???
(Hint: this works quite well as a slurm script!)
There are now 1 directory called NR.split.100 and within that there are 100 directories called NR.1, NR.2, NR.3, … NR.100.
Search against all the directories
We can now use a slurm array job to search against all of those:
#!/bin/bash
###############################################################
# #
# Search the NR using an array job. #
# #
# submit with this command: #
# #
# sbatch --array=1-100:1 ./nr_chunks.slurm #
# #
# Written by Rob Edwards. Good luck! #
# #
###############################################################
#SBATCH --job-name=NRmmseqs
#SBATCH --time=5-0
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=24
#SBATCH --mem=25G
#SBATCH -o mmseqsNRA_%a.out
#SBATCH -e mmseqsNRA_%a.err
## Memory requirement: for 100 chunks 25G (average from 72 searches: 16.3889, range: 12-20G)
set -euo pipefail
# change this to point to the output from `mmseqs createdb` on your data:
QUERY='mmseqs_formatted_data.db'
NR=$SLURM_ARRAY_TASK_ID
CHUNKS=100
mmseqs search --threads 24 $QUERY NR.split.$CHUNKS/NR.$NR/NR $QUERY.nr.$CHUNKS.$NR.db $(mktemp -d)
mmseqs convertalis --threads 24 $QUERY NR.split.$CHUNKS/NR.$NR/NR $QUERY.nr.$CHUNKS.$NR.db $QUERY.nr.$CHUNKS.$NR.m8
You can save this file as nr_chunks.slurm, edit the line with the QUERY location, and submit 100 MMSeqs2 jobs as an array job (using sbatch --array=1-100:1 ./nr_chunks.slurm). You now have 2,400 processors computing on your search, and each 24 processors is only consuming 25 GB RAM.
Note: I recommend that you adjust the number of threads, I am not sure if 24 is optimum!
We have several projects that look for all the metagenomes in the cloud, and we have several ways of searching the SRA. Here, we’ll search for all the WGS metagenomes in the SRA using a Google Big Table query.
Use SQL to find the metagenome/microbiome/metatranscriptome results
We use temporary tables to store the two main searches: what are amplicon projects and what are metagenome/microbiome/metatranscriptome projects, and then we find the projects that are metagenomes:
create temp table AMPLICON(acc STRING) as select acc as amplicon from `nih-sra-datastore.sra.metadata` where assay_type = 'AMPLICON' or libraryselection = 'PCR';
create temp table METAGENOMES(acc STRING) as select acc from `nih-sra-datastore.sra.metadata` where librarysource = "METAGENOMIC" or librarysource = 'METATRANSCRIPTOMIC' or organism like "%microbiom%" OR organism like "%metagenom%" or organism like '%metatran%';
select acc from METAGENOMES where acc not in (select acc from AMPLICON);
Then save that as a JSON file to Google Drive.
Use jq to parse the JSON file
This is probably overkill because we only have one attribute in our data.
We can edit the above SQL to get all the information about all the metagenomes. Basically, we just change the second select statement.
create temp table AMPLICON(acc STRING) as select acc as amplicon from `nih-sra-datastore.sra.metadata` where assay_type = 'AMPLICON' or libraryselection = 'PCR';
select * from `nih-sra-datastore.sra.metadata` where acc not in (select acc from AMPLICON) and (librarysource = "METAGENOMIC" or librarysource = 'METATRANSCRIPTOMIC' or organism like "%microbiom%" OR organism like "%metagenom%");
Note: In this query, the parenthesis are important to make sure we do the and and or in the right place.
Then you can export the data as a JSON Newline file to Google Drive.
Current results
At the moment, this returns 642,842 runs from the SRA
Some things we can’t find
The old study_type field that we searched (using study_type = "Metagenomics") does not appear to have mapped to bigtable.
THe old scientific name that we searched (using sample.scientific_name like "%microbiom%" OR sample.scientific_name like "%metagenom%") does not appear to have mapped to bigtable.
This is another in our series of posts on searching things in the SRA. As we noted previously, NCBI has moved most of the SRA into the clouds, which makes searching more convenient.
We can search the NCBI k-mer based taxonomy profiles too:
SELECT * FROM `nih-sra-datastore.sra_tax_analysis_tool.tax_analysis` WHERE acc = 'SRR21081434' order by ileft, ilevel
And then finally search for a record by the presence of crAssphage (taxonomy ID: 1211417)
SELECT m.acc, m.sample_acc, m.biosample, m.sra_study, m.bioproject
FROM `nih-sra-datastore.sra.metadata` as m, `nih-sra-datastore.sra_tax_analysis_tool.tax_analysis` as tax
WHERE m.acc=tax.acc and tax_id=1211417
ORDER BY m.bioproject, m.sra_study, m.biosample, m.sample_acc
LIMIT 100
(Note without the limit, this will return 140,864 records!)
But I have thousands of Accessions! What do I do?
OK, let’s take it to the next level!
We are going to create a new table in our Big Query database.
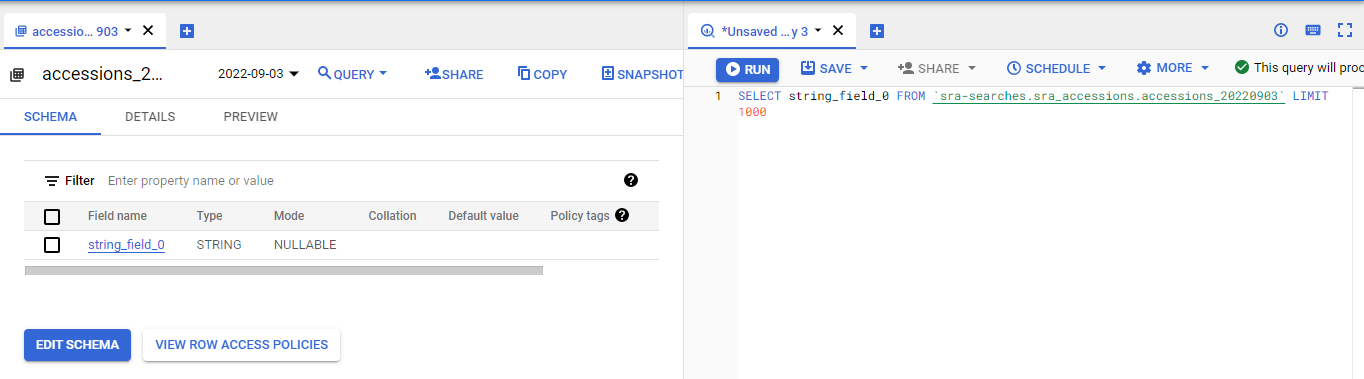
Click on your project name and choose CREATE DATA SET:
This opens a side bar where you can give your data set a name! I called my data sra_searches and because I am using the NCBI SRA data, I want to search in us (multiple regions) (although the SRA data is probably all in Iowa).
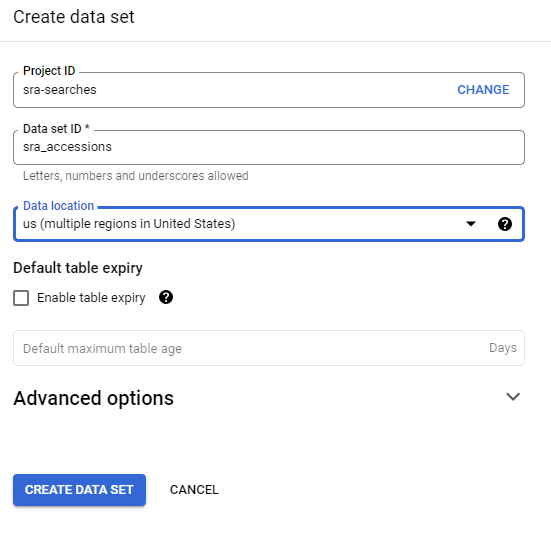
Now, expand on your data set and click on the three dots and choose Create Table
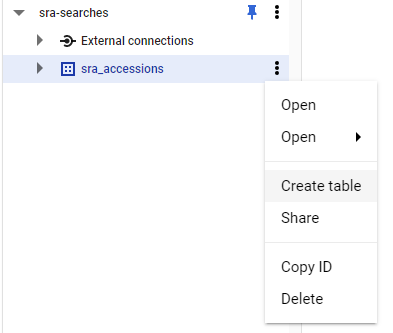
Now we need to fill in four things:
Choose Upload in the Create Table From menu
Choose your file. Use csv even if you have a single list of accessions!
Give your table a name. e.g. accessions_20220903 (because later I will have other accessions to search!)
Check the Auto-detect table format (it works well, I don’t know why it is not the default!)
This will create your table, and open it in the browser!
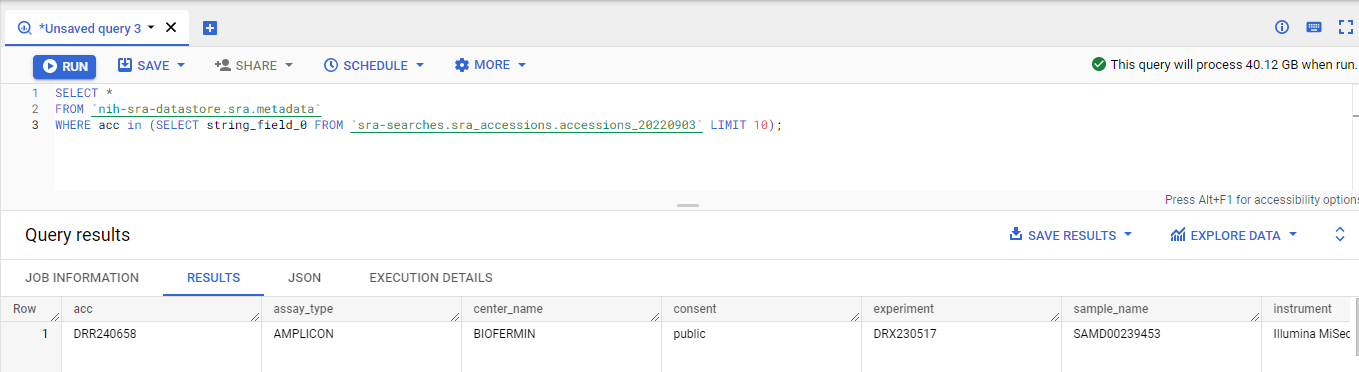
Now click on the data field that has the accessions and the query box will open to allow you to query them:
It is always therapeutic to annotate phages, but in this instance, we are specifically thinking about how we annotate phages so that we can use them for therapeutic purposes.
Rob is giving a talk at ESCMID entitled “The annotation of therapeutic phages” where he discusses some of the issues that come up. This blog post accompanies that talk and provides links to some of the papers that he discusses.
Host lifestyle prediction
It is generally accepted that lysogeny is bad for therapeutic phages, but there are ways around it. Lysogeny can lead to superinfection exclusion, recombination with other phages, and the development of phage resistance.
Here are some tools that can be used to predict whether phages are lytic or lysogenic
We can overcome lysogeny, either through engineering phages, or through Gibson Assembly based on prophage sequences. These two papers suggest some cutting edge approaches to making that happen!
But the jury is still out on how important this is! For many, espeically lytic phages, they may not care about antibiotics since they are going to kill the host anyway. There is some debate as to the importance of antibiotic resistance genes in phages.
Nonetheless, because of the overall importance of antibiotic resistance in bacterial genomes (which, after all, is the reason we are here), there are lots of databases that you can use to search for different antibiotic resistance genes.
New ways of identifying phages that have the potential for therapy are starting to emerge, and these are some of the ensemble tools that are trying to integrate multiple lines of evidence and provide support for phages for therapy.
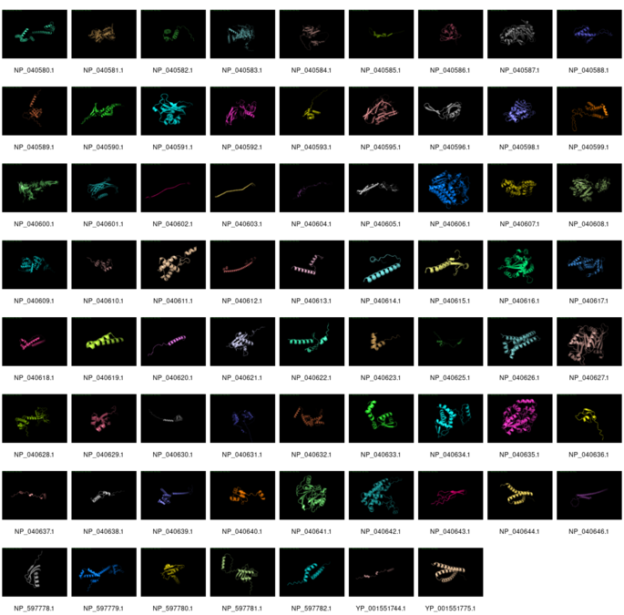
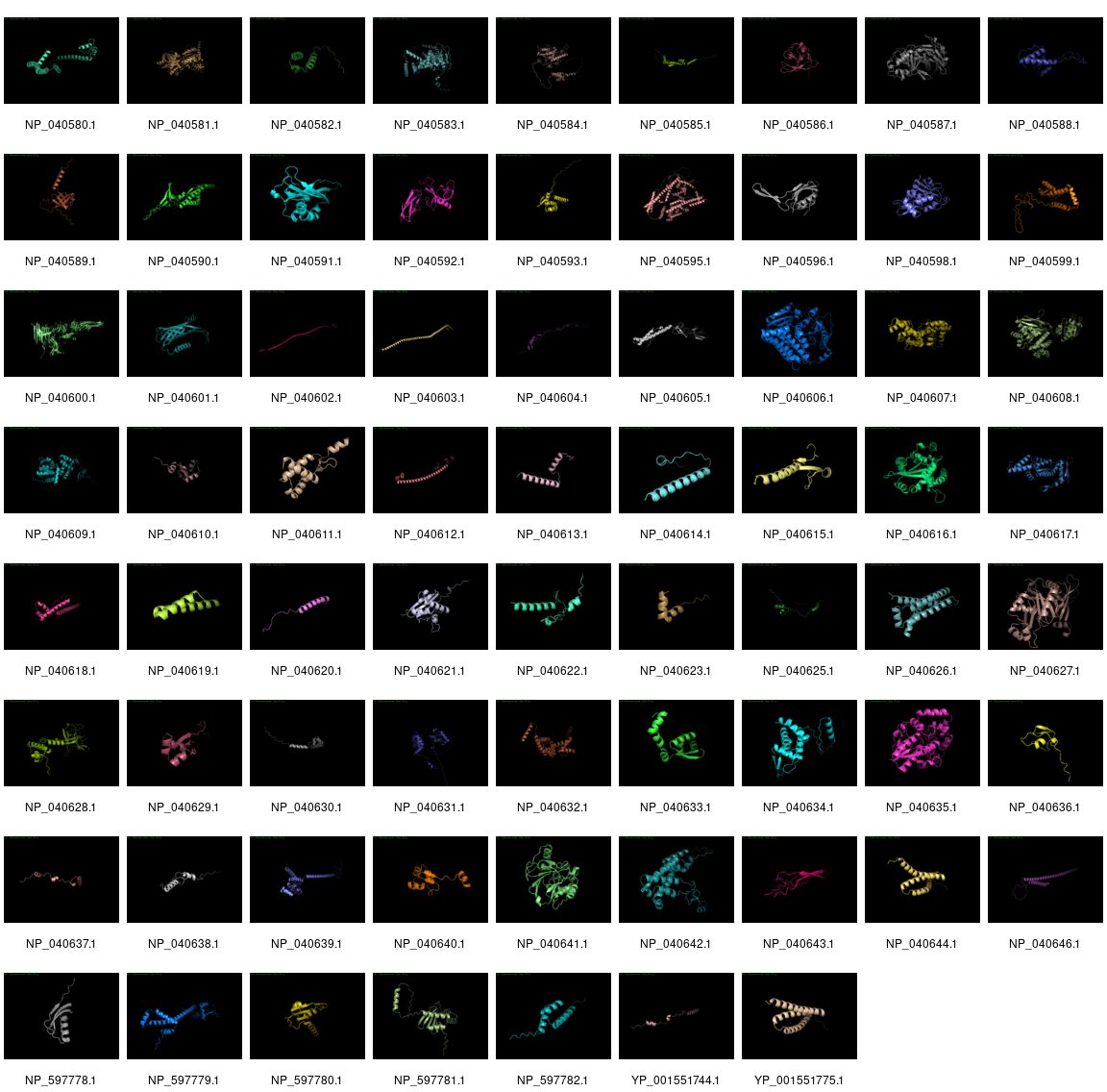
DeepMind’s AlphaFold is winning at predicting tertiary structures from primary amino acid sequences. We thought it would be fun to investigate how it performed on phage Lambda.
We took the NCBI version of λ and extracted all the proteins, and then ran them through AlphaFold. It was able to make a prediction for all the proteins except for three proteins: NP_040594.1 (144 amino acids), NP_040597.1 (232 amino acids), and NP_040645.1 (158 amino acids).
Click to see a larger version
As you can see, many of the structures are just predicted to be long alpha helices with little order, but some of the structures are complex and closer representation to the predicted structures.
There are, of course, a heap of caveats to this analysis, including the fact that we did not (at this time) filter out any of the existing phage λ structures so one would hope that those are really good!
Recently, NCBI released their new datasets API that might replace NCBI E-utils. At the moment, datasets is focused on genomes, genes, and viruses, but no doubt it will expand over time. [As an aside: I think the name is terrible, and they should use ncbi_datasets (see this tweet)]
Here is a rough guide to extracting some data about genomes using datasets.
Here are some (probably simple) mmseqs2 tips that you probably don’t remember how to do. If you need explanations or details, read the mmseqs2 wiki. Otherwise, good luck!