The immense amount of metagenomic data produced today requires an automated approach for data processing and analysis. Before any downstream analysis will be performed, the datasets should be preprocessed to ensure the quality of the data and prevent erroneous conclusions. One step of your data preprocessing (usually the last) should be to check for sequence contamination (DNA from sources other than the sample). This post will show you how to identify and remove human sequence contamination from metagenomes, but can also be applied to any other type of sequence dataset or contamination.
Sequence similarity seems to be the only reliable option to identify single contaminant sequences. However, BLAST searches against the human reference genome are slow and lack corresponding regions (gaps, variants, …). Furthermore, novel sequences were found in every new human genome sequenced. To overcome those limitations, we developed a program called DeconSeq (http://deconseq.sourceforge.net). DeconSeq allows the automated identification and removal of sequence contamination in longer-read datasets (>150 bp mean read length) using an algorithm tens of times faster than BLAST. At the moment, DeconSeq provides databases for the identification of human DNA contamination from the seven currently available human genomes. As more human genomes will be available, this reference dataset can be easily extented. If you want to find out more about DeconSeq, take a look at the FAQ and manual on its Sourceforge website.
The following step describe how to remove human sequence contaminants using the web version of DeconSeq. The sequence data can either be provided in FASTA or FASTQ format.
1. Go to http://deconseq.sourceforge.net
2. Click on “Use DeconSeq” in the top menu on the right (the latest DeconSeq web version should load)
3. Select your FASTA or FASTQ file
4. Select the retain and remove (optional) database(s)
5. Click “Submit”
Note that step 4 allows selecting two types of reference databases. The remove databases are the databases used to screen for contaminants. The retain databases are used to eliminate redundant hits with higher similarity to non-contaminant genomes (e.g. viral sequences in the human genome for viral metagenomes).
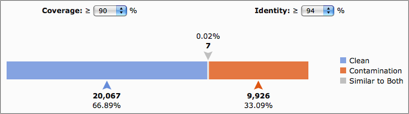
DeconSeq uses the query sequence coverage and alignment identity to identify sequences that are similar to a contaminant sequence in the remove databases. The identity is a measure for how similar the query sequence to the reference sequence is and the coverage is a measure of how much of the query sequence is similar to the reference sequence. If a retain database is selected, query sequences are classified as “Similar to Both” if they are similar to sequences in the remove and retain database using the coverage and identity thresholds. All other query sequences can be classified as “Clean” or “Contamination”, as shown below.

DeconSeq generate Coverage vs. Identity plots to guide users in their selection of threshold values. The plots show the number of matching reads for different query coverage and alignment identity values. The number of matching reads with a specific coverage and identity value defines the size of each dot in the plots. Red dots represent matching reads against the remove databases and blue dots against retain databases. The column and row sums at the top and right of each plot allow an easier identification of the number of sequences that match for a particular threshold value.

The plots for matching reads against the remove databases do not show matching reads that additionally have a match against the retain databases (A). Results for reads matching against both databases are shown in a second plot where dots for a single read are connected by lines. If the match against the remove database is more similar, then the line is colored red, otherwise blue. In B, for example, the majority of sequences is more similar to the retain databases and in C the majority is more similar to the remove databases.
If you do not require any graphical outputs or want to use your own databases, try the standalone version of DeconSeq. Note that the databases used for contamination screening have to be generated as described in the readme file distributed with the source code. The readme file also contains information on the usage of the standalone version.