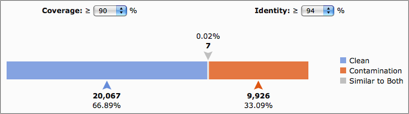
The immense amount of metagenomic data produced today requires an automated approach for data processing and analysis. Before any downstream analysis will be performed, the datasets should be preprocessed to ensure the quality of the data and prevent erroneous conclusions. One step of your data preprocessing (usually the last) should be to check for sequence contamination (DNA from sources other than the sample). This post will show you how to identify and remove human sequence contamination from metagenomes, but can also be applied to any other type of sequence dataset or contamination.