We recently released a new version of our qudaich software, designed to compare short read sequence data sets to each other. Qudaich is built around a suffix trie and provides a rapid way to compare short read data sets at the DNA or protein level. Here is how to use qudaich to compare a set of metagenomes to find out how similar they are.
Category Archives: Phage
crAssphage poops up again!
Yet again, analysis of a metagenomic sample shows that crAssphage is the most abundant phage anywhere. It also shows what a dis-service NCBI did to science by deleting the crAssphage record. We used meta-spades to reconstruct the entire crAssphage genome from someone else’s data set, but in their paper, the largest contig was ~3 kb. This analysis suggests that crAssphage is present in ulcerative colitis samples but the abundance goes down after treatment!
Getting data from the SRA
Getting data from the NCBI Sequence Read Archive is not easy. Here we combine a few of our posts to go step by step through getting the data.

How to create a phage proteomic tree
It has been a while since the original phage proteomic tree paper came out (twelve years!), and we still don’t have a web based method for doing it.
However, here are the steps and code that we use to make the phage proteomic tree.
Phage Identification
We are interested in phages — viruses that infect bacteria. For years the Edwards’ lab has been looking at new, undiscovered phages.
Recently, we identified the crAssphage, a new type of virus that has never been seen before. By looking at the sequences in metagenomes we were able to identify a set of contigs that were common among many different metagenomes. When we assembled them, they looked like a phage. We could compare them to other known phages in our database of sequences.
Working with folks in the biology department we proved that this is a circular virus by using PCR. However, we have so far been unable to culture the virus in vivo. We’re working on it, and hopefully others are too, but until that point we don’t have an image of the virus or an idea of what it does.
What is the relative biomass of crAssphage in the intestine?
Following up from the crAssphage press and comments Dan asked me the following question:
It was interesting to hear that there are 10 times as many viruses as bacteria in the body. If you have time to answer a question, I’ve always wondered about the relative biomass of bacteria compared to human cells, and now the relative biomass of viruses compared to human cells.
Inspired by XKCD’s what-if we can use some Fermi estimation to answer this. A typical virus is about 10-19 kg. (e.g. Adenovirus which is about 50kb is 2.5 x 10-19 kg [1]). A typical bacterium, like E. coli is about 10-15 kg, and a typical human cell is about 10-12 kg.
Scientists like to say that we have ~10x more bacteria than human cells and ~10x more viruses than bacteria. In the human body there are about 37 trillion cells [2] (37 x 1012, but since we are estimating we’ll round that to 1014) . Based on these estimates we have the average human weighs about 100 kg (1014 cells x 10-12 kg) in human cells, 1 kg in bacteria (1015 cells x 10-15 kg), and 0.001 kg in viruses (1016 viruses x 10-19 kg)

Phage tRNA genes
We have just published a paper describing how phages affect translation of proteins very specifically. In our case, the phage expressed a peptide deformylase with increased specificty for proteins involved in photosynthesis. That lead me to wonder how else phages affect protein synthesis, and whether they are merely trying to increase the amount of proteins being made. One way to do that might be to increase the number of tRNA-Met initiator tRNAs. To test this hypothesis I counted all the tRNAs in all the phages to see which is the most abundant. It wasn’t tRNA-Met, and after the read more I will tell you what it was.
I used tRNAScan-SE to identify all the tRNAs in all the phages in the PhAnToMe database, and from the output from tRNAScan-SE I counted all the different types. Here is a table listing all the tRNAs and their frequency in the phages:
| tRNA | Count | tRNA | Count | tRNA | Count | ||
|---|---|---|---|---|---|---|---|
| Arg | 194 | Pro | 103 | Ser | 53 | ||
| Leu | 177 | Gln | 92 | Ile | 52 | ||
| Met | 168 | Trp | 86 | His | 48 | ||
| Pseudo | 166 | Glu | 78 | Ala | 48 | ||
| Thr | 137 | Val | 77 | Tyr | 41 | ||
| Asn | 124 | Asp | 72 | Undet | 30 | ||
| Gly | 120 | Phe | 62 | Sup | 19 | ||
| Lys | 112 | Cys | 55 |
This is all of the tRNAs, and clearly there are some really big differences. A question to answer is why some tRNAs more abundant than any others?
The reference to number of phages on Earth
I have always taken (and used) for granted the 1031 number of phages in the planet. Normally, this is calculated from the estimation that there are 10 phages per prokaryotic cells, and the latter are estimated to be 1030. Usually the references to these numbers are: Jiang & Paul 1998, PMID 9687430 and Whitman 1998, PMID 9618454
Today I found what might be an older reference: Bergh et al. 1989, PMID 2755508, High abundance of viruses found in aquatic environments
Once I get access to the full-text paper (“thanks to” Nature’s unwillingness to open even older articles), I can confirm the exact phage number as claimed in 1989.
If you know of a better (aka older) reference, feel free to share it.
This number (1031), by the way, can be read as: ten nonillions (by the US numbering system)
You gotta lyse that lysin before the lysin lyses you!
Phages kill bacteria. That’s their ultimate goal. Yet, they have to maintain the bacterial cell integrity until they’re done with making new phage particles. So, they carefully control the bacterial genome till they replicate their DNA and package it in nascent phage particles. Once these are formed and are ready to leave, they need to leave. They engage in a highly timed and orchestrated procedure of poking holes in the bacterial membranes (using phage holins), degrading the bacterial peptidoglycan-based cell wall, then—if the bacterial host happens to be a gram-negative cell—breaking the outer membrane too!
In the event a phage decides to remain “dormant” inside a bacterium, things get a bit more complicated. A so-called “arms race” is generated. For bacteria, phages are time bombs that can be induced at any time to kill the bacteria. How would bacteria avoid this fatal vampirish ending? They have to “tolerate mutations” in the phage’s most dangerous protein-encoding genes. If the gene that controls phage induction is damaged, this may salvage the bacteria. Other tempting targets are the lysis modules! If lysins or holins are disabled, the domant prophages may remain captive forever (or rather until prince “helper phage” comes and frees them from that peptidoglycan-walled prison.
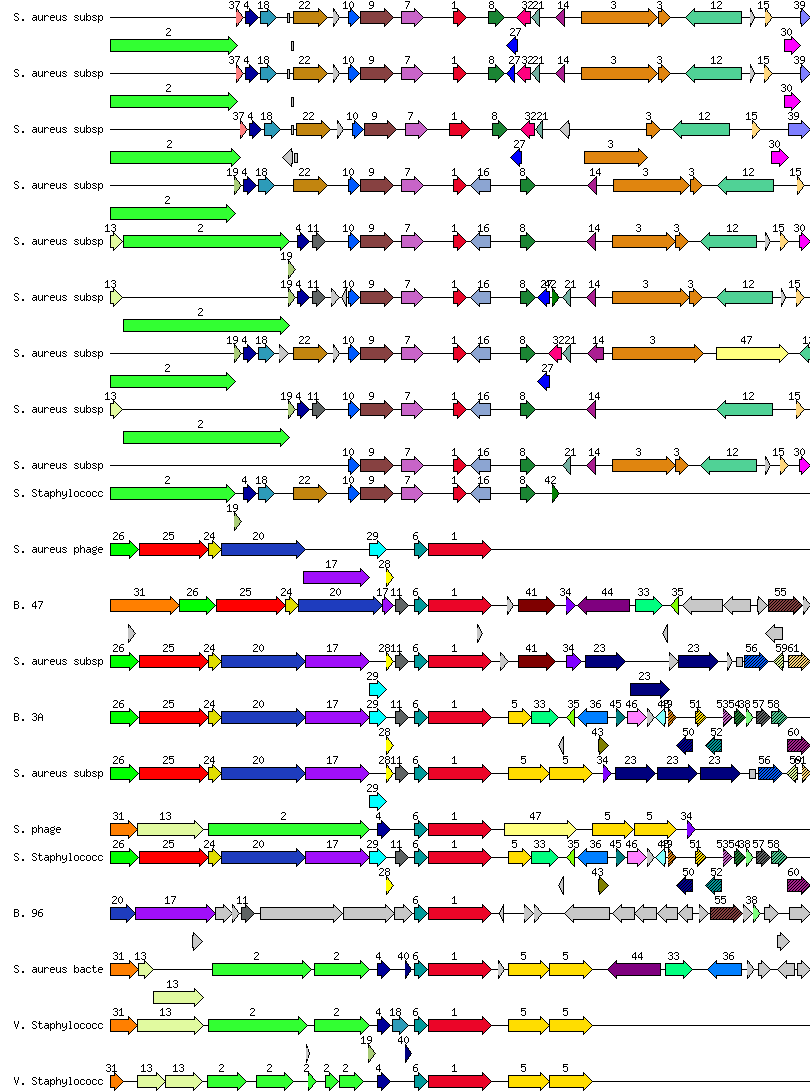
So, if you’re a bacterium, it’s smart to disable the lysin genes, one way or another. If you’re a scientist studying bacterial and phage genomes, there is no better way to find this out than using the subsystems-based SEED server. Using subsystems allows you to find out how closely related phages and prophages may have very different lysin genes. In the diagram below, a bunch of staphylococcal phage and prophage genomes are compared. You will notice immediately how some of their lysins (in Red, labeled # 1) are sometimes truncated. A truncated lysin is bad news for a phage. It means the phage is on its way to be enslaved by the bacterium for long years to come!
Identifying Prophages in Bacterial Genomes
Finding prophages in microbial genomes remains a problem with no definitive answer. The majority of existing tools rely on detecting genomic regions enriched in proteins with known phage homologs, which hinders the de novo discovery of phage regions. In this study, a weighted phage detection algorithm, Phage_detector was developed based on seven distinctive characteristics of prophages i.e. protein length, transcription strand directionality, customized AT and GC skew, the abundance of unique phage words, phage insertion points and the similarity of phage proteins. The first five characteristics are capable of identifying prophages without any sequence similarity with known phage genes. Phage_detector locates prophages by ranking genomic regions enriched in distinctive phage traits, which leads to the successful prediction of 92% of prophages (including 33 previously unidentified prophages) in 95 complete bacterial genomes with 8% false negative and 18% false positive.